
Zhisong Jeff HE
何志嵩
RESEARCH INTEREST
As a computational biologist, I'm mostly interested in figuring out how complex primary biological systems, e.g. human brains, evolve and develop with quantitative methods. The understanding of primary biological systems can be next transferred into the capacity of engineering systems. Bioengineering, especially cell type engineering, is therefore my another interest.
My current focuses include:
-
understanding early human brain development, by high-throughput genomic profiling and perturbations in organoids
-
developing computational toolkit to integrate multi-experiment and multi-modal single-cell and spatial genomic data
-
developing computational models to describe and predict changes of cell type identities
HIGHLIGHTED PROJECTS
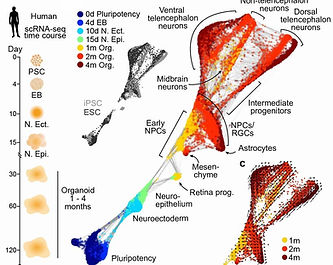
Integrated transcriptomic cell atlas of human neural organoids
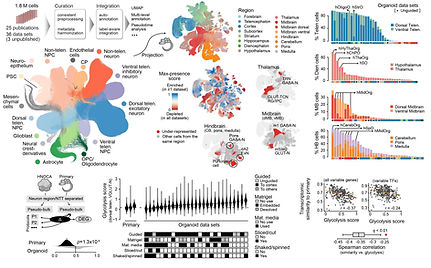
We are interested in how different neural organoid protocols performed and how similar are cells generated in neural organoids to their primary counterparts. Therefore, we integrated 1.8 million cells from 36 different scRNA-seq data sets covering 26 different neural organoid protocols, to generate the first draft of human neural organoid cell atlas (HNOCA). To compare to the primary developing human brains, we re-analyzed the recent first-trimester developing human brain cell atlas (Braun et al. 2023), and projected HNOCA to the primary atlas to 1) refine regional annotation of HNOCA, 2) evaluate over-/under-representation of primary cell states in organoids, and 3) perform transcriptomic comparison between matched neural cell types in organoids and primary tissues. We observed different neural cell types in primary that are under-representated in any current organoid protocols, and identified glycolysis as a universal cell stress signature. Experimental procedures related to minimizing the stress signature, which was shown to have minimal effect to cell type core identities, were identified.
Spatial-temporal multimodal phenotyping of human retinal organoids
We implemented the iterative indirect immunofluorescence imaging (4i) technique on human retinal organoid (hRO) tissues, to measure >50 proteins on the same tissues and obtained the spatial-temporal atlas of hRO development. Meanwhile, we also generated scRNA-seq, scATAC-seq and scMultiome data of hRO development, and developed the integration procedure to generate the spatial-temporal tri-modal (epigenome-transcriptome-proteome) atlas of hRO development. Using the atlas, we analyzed the spatial distribution of different cell types across the development, and investigated the regulatory mechanisms controlling the cell type differentiation by reconstructing the gene regulatory network. We further applied CROP-seq experiment to investigate the regulatory functions of multiple selected transcription factors, highlighting OTX2 as a significant regulator.

The paper has been published in Nature Biotechnology in 2023. The earlier version is available at biorxiv.
Cell fate regulome of brain organoid development
Cerebral organoids combined with single-cell genomic technologies provide opportunities to explore gene regulatory networks (GRNs) underlying human brain development. Here we generated single-cell transcriptomic (scRNA-seq) and epigenomic (scATAC-seq) over a dense time course covering multiple phases of brain organoid development. We identified temporally dynamic and brain region-specific regulatory regions. We further developed Pando, a regression-model-based GRN inference method incorporating multi-omic data and transcription binding site predictions, and applied the method to infer a global GRN describing organoid development.
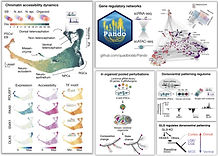
We also used pooled genetic perturbation with scRNA-seq readout to assess transcription factor requirement for cell fate and state regulation in organoids, and identified GLI3 being required for cortical excitatory neuron fate establishment in humans. The further single-cell multiome experiment on GLI3-mutant organoids, together with the Pando-inferred GRN, identified a regulome central to the dorsoventral telencephalic fate decision. We identified HES4/5, two famous Notch effectors, were strongly and directly regulated by GLI3 in a Notch-independent manner. Altogether, we provide a framework for how multi-brain region model systems and single-cell technologies can be leveraged to reconstruct human brain developmental biology.
The paper has been published in Nature in 2022. Its original version was preprinted in biorxiv in 2021.
Lineage recording reveals dynamics of cerebral organoid regionalization
Diverse regions develop within cerebral organoids generated from human induced pluripotent stem cells (iPSCs), however it has been a challenge to understand the lineage dynamics associated with brain regionalization. Here we establish an inducible lineage recording system that couples reporter barcodes, inducible CRISPR/Cas9 scarring, and single-cell transcriptomics to analyze lineage relationships during cerebral organoid development. We infer fate-mapped whole organoid phylogenies over a scarring time course, and reconstruct progenitor-neuron lineage trees within microdissected cerebral organoid regions. We observe increased fate restriction over time, and find that iPSC clones used to initiate organoids tend to accumulate in distinct brain regions. We use lineage-coupled spatial transcriptomics to resolve lineage locations as well as confirm clonal enrichment in distinctly patterned brain regions. Using long term 4-D light sheet microscopy to temporally track nuclei in developing cerebral organoids, we link brain region clone enrichment to positions in the neuroectoderm, followed by local proliferation with limited migration during neuroepithelial formation.
The paper has been published in Nature Methods in 2021, and it is also available in biorxiv.

Integration of single-cell genomic data using similarity spectrum representation
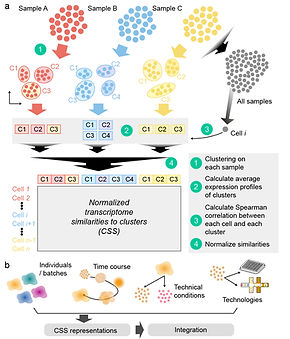
Recent advances of single-cell genomic technologies, especially scRNA-seq, offer great capacity to resolve heterogeneity of cell types and cell states in biological samples. However, current scRNA-seq technology only provides a snapshot on limited samples at one time. Joint analysis on many samples across multiple experiments and different conditions is often required, when the biological variation of interest is confounded by other technical factors. Several computational integration methods have been developed to address some of these issues. However, benchmarking on these integration methods have revealed that each method presents different performance in various scenarios. More options are still in need.
Here, we propose an unsupervised scRNA-seq data representation namely Cluster Similarity Spectrum or CSS. It considers every cell cluster in each sample for integration as an intrinsic reference, and represents each cell by its transcriptome similarities to clusters across samples. We apply CSS to integrate cerebral organoid data from different human iPSC lines, batches, modalities, and conditions. We show that technical variation caused by experimental conditions or protocols can be largely reduced with the CSS representation, and CSS has similar or even better performance compared to other integration methods.
The manuscript has been published in Genome Biology in 2020, and also preprinted in biorxiv. The script is available in GitHub as an R package simspec.
Evolution of primate brain developmental trajectory using single-cell RNA-seq and cerebral organoids
To better understand the biological foundation of the differences of cognitive ability between humans and other non-human species relatives, e.g. chimpanzees and macaques, one would have to understand the developmental divergences among those species, as the changes of the brain developmental trajectories may largely contribute to the structural and functional differences between humans and other primate species. While it is hardly feasible to collect a large number of fetal brain samples from different primate species including humans, the emergence of 3D brain cell culture technique, which generates brain organoids that have been proven to recur many features of real brain development, provides a great changes to compare primate brain developmental trajectories on the molecular levels in a more controlled and large-scaled manner.
In this project, we applied single-cell RNA-seq to capture the heterogeneity and complexity of cerebral organoids, to investigate divergence of different cell types along their developmental trajectories between humans and non-human primates. We observed timing differences of neuron differentiation and maturation between human and chimpanzee cerebral organoids, with human neurons maturing significantly slower. Independent from that, hundreds of genes were found being differentially expressed between human and chimp along cortical excitatory neuron development. In addition, we generated single-nucleus RNA-seq data from adult prefrontal cortex in humans, chimps and macaques. While the neuronal universal and subtype-dependent signatures are consistent in adult and cerebral organoids, we observed different human-specific DEs in the two system, which are found to be relevant to the developmental stage relevant transcriptome programs, suggesting the complementarity between the in vitro cerebral organoid system and the in vivo adult primary tissues when studying brain evolution.
The manuscript has been published in Nature in 2019. It is also available in biorxiv (longer version with more details). Data visualization and exploration is accessible via online ShinyApp scApeX. Important scripts for analysis are available in GitHub. Raw data are archived in ArrayExpress, and parts of the processed data are archived in MendeleyData (DOI: 10.17632/z4jyxnx3vp).
Transcriptome of primate cortical laminae and its evolution in primates
The comparison of different primates provided a lot of interesting information. Significantly more human-specific changes, especially those with uneven distribution across layers, were identified comparing with the chimpanzee-specific changes, suggesting an accelerated evolution in the human lineage which implies the potential positive selection during human evolution. The layer specificity transition of gene expression from L5 to L3, which largely represented changes of the neuron population, was particularly of great interest, as its frequency is significantly higher in humans, and the data suggested that it was a continuous process happened since the separation of primate and rodent ancestors. The transcriptome comparison also suggested that glial cells, especially astrocytes, largely contributed to the human-specific changes, and may play an more important role in the generation of human-specific cognitive ability than people thought before.
The paper has been published in Nature Neuroscience in 2017.

In mammalian brain, there is a six-layer structure with each layer named from L1 to L6. These layers are characterized by their distinct cell type constitution and neuronal connectivities, which results in different functions and roles. However, the molecular signatures and evolutionary properties of these layers, especially those in human brains, are largely unknown. In this study, we designed the unsupervised sectioning method to generate cortical sections which can reflect the cortical layers, and used RNA-seq to generate the transcriptome atlas of the primate cortical laminar structure.